Este documento se preparó para promover los objetivos educativos de La Fundación del Cáncer Carcinoide (Carcinoid Cancer Foundation) e informarle sobre la existencia y las características del cáncer neuroendocrino. Mientras la información contenida en este documento representa información actualizada sobre el cáncer neuroendocrino, esta información no debe utilizarse como sustituto de una visita con su médico si hay alguna duda sobre su salud.
Con el más profundo agradecimiento a la Dra. Jaydira Del Rivero, Directora de la Clínica de Tumores Raros/Programa de Tumores Neuroendocrinos del Instituto Nacional del Cáncer, por traducir la Revisión del Cáncer Neuroendocrino al español.
Esta reseña del Cáncer Neuroendocrino fue escrita inicialmente por Richard R.P. Warner, MD, especialista en cáncer neuroendocrino y fundador de La Fundación del Cáncer Carcinoide (Carcinoid Cancer Foundation). Agradecemos las contribuciones del Dr. Satya (Nanu) Das, MD, MSCI, Líder del Programa de Tumores Neuroendocrinos en Vanderbilt-Ingram Cancer Center en Nashville, Tennessee, quien actualizó la Revisión en el 2021. Con agradecimiento también a Edward M. Wolin, MD, Director del Centro de Tumores Carcinoides y Neuroendocrinos del Hospital Mount Sinai de Nueva York, por su ayuda con la Revisión.
Introducción y conceptos básicos
Primero debe comprender algunos conceptos básicos sobre el cuerpo humano y cómo se desarrollan y crecen los tumores. Cada parte del cuerpo, desde la piel hasta el corazón, los músculos, las glándulas y todos los demás órganos, está compuesta de células microscópicas al igual que los ladrillos que forman la estructura de un edificio. Pero a diferencia de los ladrillos de construcción, las células del cuerpo se forman en clases especializadas en apariencia, estructura y función para los propósitos del órgano o la parte que forman. Además, a diferencia de los ladrillos de un edificio, que una vez formados y colocados no cambian durante toda la vida del edificio, las células vivas del cuerpo se degeneran constantemente, se desgastan y se regeneran / reemplazan por células idénticas. Este proceso de replicación ocurre continuamente y está regulado por controles complejos genéticos y hormonales tanto dentro de las células individuales como por influencias de otras partes del cuerpo. Cuando algo sale mal con este sistema regulador delicado y complejo, la replicación celular a veces se produce de manera no regulada, lo que resulta en un crecimiento tumoral (neoplasia). Si este crecimiento excesivo es algo limitado y no se disemina a otras áreas ni amenaza con exprimir o reemplazar estructuras adyacentes, se considera un tumor benigno; es decir, no amenaza la vida. Sin embargo, si el crecimiento es más agresivo y amenaza los tejidos circundantes o envía “plántulas” (metástasis) para crecer en áreas distantes, entonces tiene el potencial de ser fatal y se considera maligno; eso significa que es un cáncer.
Hay algunos tipos de crecimientos que se encuentran a medio camino entre estas dos clasificaciones de benignos y malignos. Los tumores neuroendocrinos (TNE) son los que ocurren con más frecuencia de estos tipos raros de crecimientos “intermedios”. Se les ha llamado “cánceres en cámara lenta” porque, aunque los TNE tienen el potencial de hacer metástasis y ser fatales, tienden a crecer tan lentamente que las personas diagnosticadas con estos tumores suelen vivir muchos años y, a veces, una vida normal. La amplia variedad de tratamientos ahora disponibles hace que las perspectivas para la mayoría de los pacientes con TNE más agresivos sean más esperanzadoras de lo que solían ser, pero hablaremos de esto más adelante.
Tumores neuroendocrinos: ¿qué son? ¿Benigno o maligno?
Los tumores neuroendocrinos son relativamente recién llegados al reconocimiento médico. Se identificaron por primera vez como un tipo específico de crecimiento y distinto a mediados del siglo XIX, y el nombre “carcinoide” se aplicó por primera vez en 1907 por el Dr. Siegfried Oberndorfer en Europa en un intento de designar estos tumores como a medio camino entre carcinomas (cánceres) y adenomas (tumores benignos). En años más recientes, los tumores carcinoides han llegado a referirse específicamente a los TNE que se originan fuera del páncreas.
Se encontró que los tumores neuroendocrinos surgen de las células del Sistema Neuroendocrino Difuso, células enterocromafinas (células glandulares productoras de hormonas endocrinas) distribuidas ampliamente en el cuerpo pero que se encuentran con mayor frecuencia en los pulmones y luego, con una frecuencia decreciente, en el intestino delgado, el recto, apéndice y páncreas. Con mucho menos frecuencia, estas células pueden estar ubicadas en los ovarios, los testículos, el hígado, los conductos biliares y otros lugares. Estas células tienen características especiales y peculiares que las hacen identificables bajo el microscopio. Se tiñen de una manera especial cuando se ponen en contacto con productos químicos que contienen plata. Las tinciones especiales para las hormonas especificas que pueden producir las células enterocromafinas, identificarán las sustancias hormonales en las células tumorales carcinoides y, por lo tanto, confirmarán el diagnóstico del examen microscópico en los TNE con biopsia.
Una de las características más importantes de un TNE es el grado que se le asigna. El grado lo realiza un patólogo y, por lo general, es más importante para los TNE de bajo grado (todos los carcinomas neuroendocrinos poco diferenciados son, por definición, de alto grado). Para los TNE gastrointestinales y pancreáticos, a los tumores se les asigna un grado basado en el índice mitótico (qué tan rápido se dividen las células bajo el microscopio) y el índice Ki-67 (otra medida de la replicación celular). Para los TNE de pulmón, los tumores se les asigna un grado basado en el índice mitótico.
Solo en 1954 se describió y aceptó por primera vez el síndrome carcinoide como una entidad patológica específica. Thorsen, Biorck, Björkman y Waldenstrom, un grupo de médicos de Estados Unidos y Escandinavia, reconocieron por primera vez la naturaleza de los diversos síntomas asociados con algunos TNE que se conocen como síndrome carcinoide y lo describieron en una revista médica. Este síndrome, que se comentará con mayor detalle más adelante, consiste en un grupo de síntomas y hallazgos en la exploración física y de laboratorio que son causados ??por las potentes sustancias producidas por los tumores neuroendocrinos.
A principios de la década de 1990, el desarrollo y la disponibilidad del octreótido (Sandostatin) por parte de la compañia farmacéutica Sandoz (ahora Novartis) proporcionó un fármaco importante para el tratamiento del síndrome carcinoide y para los TNE en general. Este fármaco se deriva de la hormona somatostatina de origen natural. En Europa, Estados Unidos y algunos países de otras partes del mundo también se utilizan otros análogos de la somatostatina como lanreotida (Somatuline Depot) (Ipsen).
Estadísticas
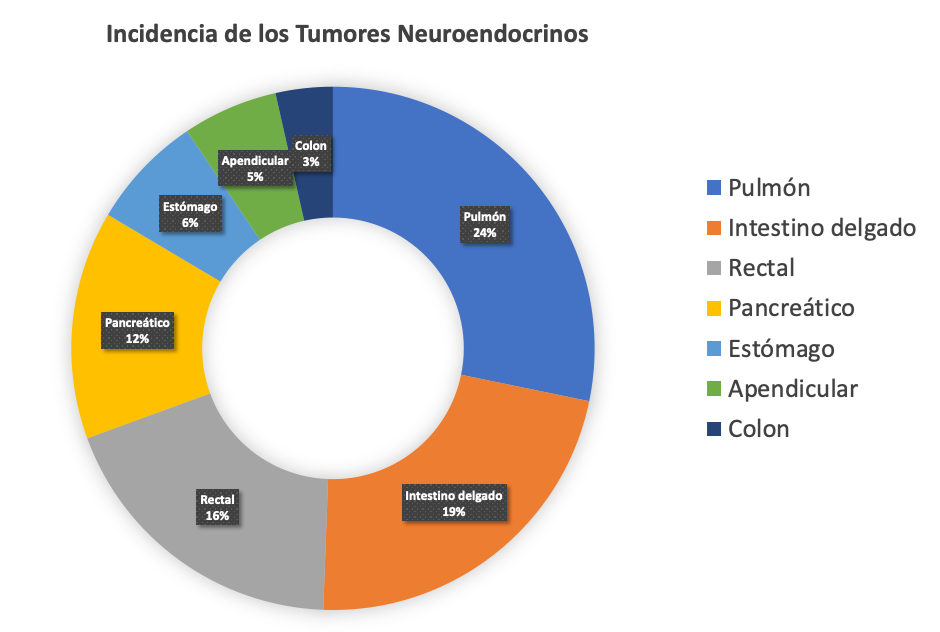
La incidencia (nuevos casos por año) y la prevalencia (número total de pacientes con cáncer) de los TNE ha aumentado de manera constante durante las últimas 4 décadas. En 2012, hubo 7 casos de NET por cada 100,000 personas en los Estados Unidos. Comparativamente, en 1973, este número era de 1 caso (s) por 100,000. Casi el 50% de este aumento se puede atribuir al hecho de que más pacientes se someten a tomografías computarizadas o resonancias magnéticas y que la calidad de estas exploraciones ha mejorado significativamente. Además, el conocimiento de los TNE en la comunidad médica y el público en general ha aumentado significativamente. Sin embargo, las otras razones de este aumento aún no se comprenden bien. La incidencia de tumores neuroendocrinos varía según el origen del tumor primario. De mayor a menor, la incidencia de TNE sigue el siguiente orden (Figura 1):
- pulmón – 24%
- intestino delgado – 19%
- rectal – 16%
- pancreático – 12%
- estómago – 6%
- apendicular – 5%
- colon – 3%
También hay algunos lugares extremadamente raros de los que pueden surgir los TNE y que se pueden diseminar e incluyen la vesícula y los conductos biliares, los ovarios, los testículos, la vejiga urinaria, la próstata, la mama, los riñones, el cerebro. (generalmente solo se observa en casos de carcinoma neuroendocrino de alto grado) y la glándula del timo y, en algunos casos mucho mas raros, el ojo y el oído (Figura 2).
Debido a que muchos pacientes viven durante muchos años con su enfermedad, los TNE que surgen en el tracto gastrointestinal y el páncreas constituyen la segunda categoría más prevalente de cáncer gastrointestinal después del cáncer color rectal. En cuanto al estadio de la enfermedad, en el momento del diagnóstico los pacientes presentan enfermedad localizada, regional (con afectación de los ganglios linfáticos) y a distancia en el 50%, 20% y 30% de los casos, respectivamente.
La segunda ubicación más común de la que surgen los TNE es el intestino delgado. Los tumores de cualquier tipo en el intestino delgado son raros y comprenden solo el 1% de todos los cánceres del tracto gastrointestinal. Sin embargo, los TNE comprenden aproximadamente el 50% de todas las neoplasias malignas del intestino delgado. Su tamaño cuando se diagnostica por primera vez es muy importante, ya que la probabilidad de que ya se haya propagado es directamente proporcional a su tamaño. Si el tumor tiene más de 2 cm de diámetro (casi 1 pulgada), las posibilidades de diseminación son superiores del 50%. Inicialmente, el tumor simplemente crece hacia la pared del intestino desde el revestimiento intestinal donde comienza. Sin embargo, eventualmente puede atravesar la pared y luego extenderse a los ganglios linfáticos cercanos, los canales linfáticos y los vasos sanguíneos y luego puede extenderse a lugares más distantes como el hígado, el peritoneo, los huesos, los pulmones e incluso el corazón.
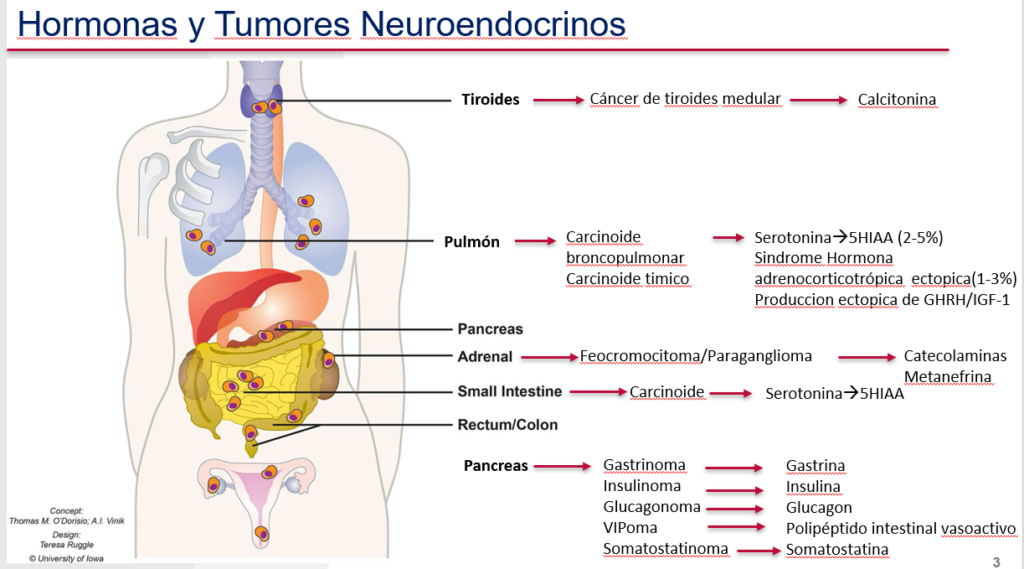
Figure 2. Tumores neuroendocrinos y las hormonas que pueden producir
TNE del intestino delgado
Las estadísticas sugieren que el 1% de la población desarrollará TNE diminutos y médicamente insignificantes en algún momento de su vida, con mayor frecuencia en el tracto intestinal. Por razones que no se comprenden completamente, en algunas personas estos TNE crecerán y se volverán diagnosticables. Los TNE del intestino delgado constituyen el 50% de todos los cánceres de intestino delgado. Estos tumores se originan en la pared del intestino delgado. Desafortunadamente, debido a las dificultades para hacer un diagnóstico temprano, hasta la mitad de estos pacientes pueden tener enfermedad metastásica en el momento en que inicialmente se les diagnostica. Aproximadamente el 50% de los pacientes con TNE del intestino delgado desarrollarán diseminación distante (metástasis) y aproximadamente 1/3 de los que se han diseminado desarrollarán síndrome carcinoide (esto se discutirá más adelante en este artículo).
TNE del estómago (TNE gástricos)
Los tumores neuroendocrinos que se originan en el estómago se clasifican de manera diferente a otros TNE. Aparte de que se les asigne un grado, estos NET también se clasifican en tres tipos:
Tipo 1. Estos se asocian con anemia perniciosa u otras afecciones que causan la degeneración del revestimiento del estómago con pérdida de la producción normal de ácido gástrico. Suelen ser múltiple, de tamaño pequeño e incluso de tamaño microscópico. Con poca frecuencia se diseminan y, por lo tanto, tienen muy buen pronóstico. La extirpación quirúrgica de la porción final del estómago (antro) generalmente detiene la producción de la hormona gastrina y puede provocar que disminuyan el tamaño de estos tumores. En algunos casos, también se pueden tratar con análogos de somatostatina como octreotida o lanreotida.
Tipo 2. Algunos carcinoides gástricos pueden aparecer como parte del síndrome de neoplasia endocrina múltiple tipo 1 (NEM1). Suelen ser de crecimiento lento y están asociados con otros tumores de las glándulas endocrinas en el mismo individuo. Debido a que producen la hormona gastrina, a menudo causan múltiples úlceras en el estómago y en el duodeno y también puede provocar diarrea.
Tipo 3. Los carcinoides gástricos de tipo 3 surgen en el estómago sin ninguna predisposición especial, al igual que la mayoría de los carcinoides del tracto intestinal. Pueden crecer hasta alcanzar un tamaño grande y aproximadamente el 50% de los casos se habrán diseminado fuera del estómago. Pueden causar malestar o sangrado y deben manejarse de manera más agresiva.
TNE Apendicular
Un TNE apendicular (appendiceal NET) ocurre con mayor frecuencia en la punta del apéndice. Aproximadamente el 50% de todos los tumores de apéndice son TNE. Este tipo de tumor neuroendocrino generalmente no causa síntomas y, con mayor frecuencia, se encuentra de manera incidental en el momento de la apendicectomía. Un TNE apendicular que se limita al apéndice tiene una alta probabilidad de éxito en el tratamiento con cirugía. Los casos tempranos pueden tratarse con una simple apendicectomía laparoscópica, aunque algunos casos más avanzados requieren una cirugía más compleja. De todos los TNE, los que surgen en el apéndice tienen menos probabilidades de hacer metástasis (diseminación). Cuando el tumor mide al menos o mas de 2 centímetros o invade el tejido llamado mesoapéndice, una simple apendicectomía no es suficiente y se necesita un procedimiento quirúrgico más grande (llamado hemicolectomía derecha). De todos los TNE, los que surgen en el apéndice demuestran el comportamiento más indolente o benigno. Los pacientes con TNE apendiculares muestran una diseminación distante muy rara y el 87% de las personas con tumores que se extirpan quirúrgicamente están vivas después de 5 años. Un TNE se encuentra generalmente por accidente en 1 de cada 200 a 300 apéndices extraídos en la cirugía.
TNE rectales
El segundo menos maligno de estos tumores son los TNE rectales. Los pacientes con estos tumores muestran una tasa de supervivencia a 5 años de más del 70%.
TNE de pulmón
El pulmón es la ubicación más común donde surjan los TNE. Los TNE de pulmón a menudo se asocian con sus propias peculiaridades especiales, enfoques de diagnóstico y formas de tratamiento. Se puede encontrar un excelente resumen de este tema en el sitio web de la Sociedad Estadounidense del Cáncer: Información sobre el tumor carcinoide de pulmón. American Cancer Society’s website: Lung Carcinoid Tumor Information.
¿Qué es el síndrome carcinoide?
Las células del TNE pueden producir muchos tipos de hormonas y péptidos. Los pacientes que tienen tales tumores se dicen que tienen tumores neuroendocrinos “funcionales”. Los TNE del intestino delgado son el tipo de tumor TNE que secreta hormonas con mayor frecuencia. Los TNE que producen grandes cantidades de hormonas y otras sustancias químicas potentes pueden causar enrojecimiento de la cara, el cuello y el torso, diarrea acuosa y ataques de sibilancias similares al asma. Estos síntomas causan el síndrome carcinoide. Los episodios del síndrome carcinoide pueden ser muy poco frecuentes al principio, pero gradualmente pueden ocurrir con más frecuencia y, cuando son lo suficientemente graves, pueden asociarse con presión arterial baja, dificultad para respirar y desmayos. El alcohol o el estrés (físico o emocional) a veces provocan ataques, pero también pueden ocurrir de forma espontánea. Después de un tiempo, el enrojecimiento puede volverse persistente en algunas personas. La diarrea también puede volverse crónica y acompañada con pérdida de peso. Más allá de los síntomas por sí solos, las hormonas pueden causar daño a las válvulas cardíacas (más comúnmente válvulas cardíacas del lado derecho) u otras alteraciones cardíacas, como ritmos cardíacos anormales. La enfermedad de la válvula carcinoide en última instancia puede resultar en insuficiencia cardíaca. La enfermedad de la válvula carcinoide, cuando es significativo, pone en peligro la vida al menos que se reemplacen las válvulas enfermas. En algunos casos, los síntomas del síndrome carcinoide que resultan de las hormonas y sustancias químicas producidas por el tumor, son peores que los síntomas del crecimiento del tumor en sí (Figura 3).
En raras ocasiones, el síndrome carcinoide también puede ser el resultado del TNE del pulmón, los ovarios y otros órganos.
Todavía no está del todo claro cuál de las sustancias es responsable de cada uno de los síntomas del síndrome carcinoide. Sin embargo, casi todos estos tumores producen serotonina y bradicinina. Otras sustancias cuyos nombres puede encontrar a veces en relación con estos tumores y que a menudo se producen en asociación con tumores neuroendocrinos son: sustancia P, pancreastatina, neurotensina, polipéptido pancreático, neuroquinina A, motilina y hormona natriurética auricular (ANH), así como otras hormonas peptídicas (Figura 2).
Los síntomas del síndrome carcinoide se pueden tratar eficazmente mediante el uso de análogos de la somatostatina (octreótido o lanreótido), un fármaco anti-serotonina conocido como telotristat etilo o mediante la reducción del volumen de metástasis en el cuerpo (mediante cirugía, terapia dirigida al hígado, terapia con radionúclidos con receptores de péptidos [PRRT] u otros tratamientos sistémicos contra el cáncer). Otros TNE pueden producir hormonas diferentes y, por lo tanto, causar otros síndromes además del síndrome carcinoide. Esto es relevante porque los tumores neuroendocrinos pancreáticos generalmente no secretan serotonina, pueden secretar otras hormonas como el glucagón, gastrina, insulina o polipéptido intestinal vasoactivo (VIP). Estos causan los siguientes síndromes: síndrome del glucagonoma, síndrome de Zollinger-Ellison, insulinoma y cólera pancreático de Verner-Morrison WDHA. Lea más aquí: https://bit.ly/endocrinesyndromes.
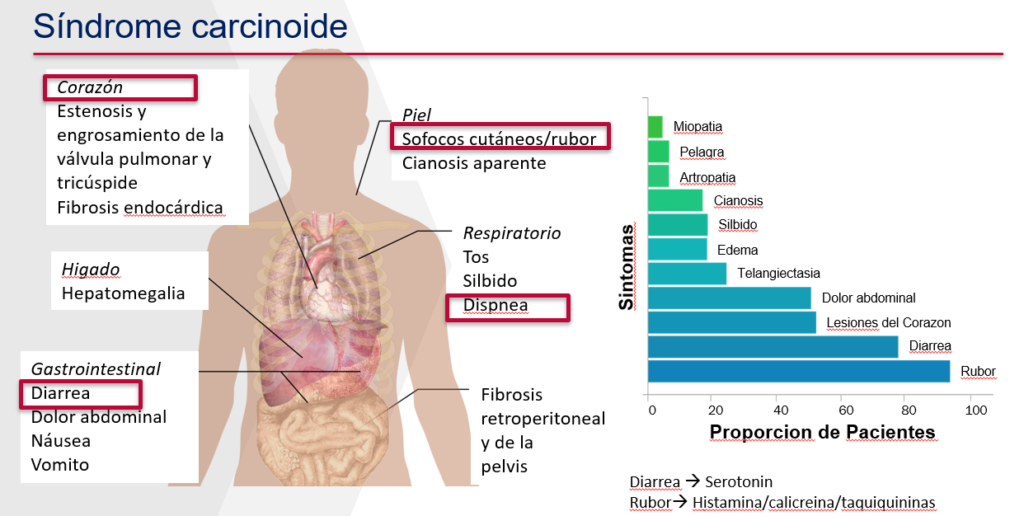
Figure 3. Síntomas del síndrome carcinoide
Diagnóstico
Los TNE que no son funcionales pueden crecer silenciosamente durante muchos años entre el inicio de cualquier síntoma y su diagnóstico. Dependiendo del sitio del tumor original o primario, algunos síntomas pueden incluir dolor abdominal inexplicable, hinchazón en el lado derecho del abdomen, distensión/ firmeza and plenitud abdominal. Por lo general, en ese momento, se realiza una tomografía computarizada o una resonancia magnética que sugiere un proceso canceroso. Sin embargo, el diagnóstico se establece típicamente mediante biopsia.
Los TNE funcionales a menudo se diagnostican antes, especialmente si liberan hormonas que producen síntomas del síndrome carcinoide u otros síndromes. El diagnóstico del síndrome carcinoide en general se puede confirmar rápidamente haciendo una prueba de 5-HIAA en plasma (sangre) o 5-HIAA en orina de 24 horas. 5-HIAA significa ácido 5-hidroxiindolacético, que es el principal producto de desglose (desecho) de la serotonina. Aunque anteriormente la prueba de orina de 24 horas era el único medio de cuantificar 5-HIAA, una prueba más reciente basada en sangre ha demostrado la misma capacidad de detección. Ciertos alimentos y medicamentos deben evitarse uno o dos días antes y el día de la recolección de orina, ya que pueden causar resultados falsos en las pruebas. Estos incluyen: plátanos, piña y su jugo, ciruelas rojas, aguacate, nueces y otras nueces, kiwi, tomates, varios medicamentos para la tos, medicamentos relajantes musculares, acetaminofén (Tylenol), cafeína, fluorouracilo, soluciones de yodo (solución de Lugol), fenacetina, inhibidores de MOA (ciertos antidepresivos fármacos), isoniazida y fenotiazina (Compazine, Thorazine). Para obtener más información sobre cómo prepararse para una prueba de orina de 24 horas, haga clic aquí. how to prepare for a 24-hour urine test click here.
Además de las tomografías computarizadas o resonancias magnéticas iniciales que a menudo detectan el tumor neuroendocrino como se describió anteriormente, es importante que los pacientes después se sometan a imágenes basadas en el receptor de somatostatina. Anteriormente, las imágenes basadas en el receptor de somatostatina se realizaban a través de un octreoscan. Ahora, el Octreoscan ha sido reemplazado en gran medida por la tomografía de emisión de positrones PET con dotatate (galio-68 o cobre-64). El PET dotatate o PET-Galio68 puede detectar muchas más lesiones en comparación con el Octreoscan, casi como una televisión de alta definición frente a una televisión de definición estándar (Figura 4). Si están disponibles, las tomografías por emisión de positrones con dotatate siempre deben realizarse en pacientes con tumores neuroendocrinos recién diagnosticados. Las razones para someterse a un PET con dotatate son dos: primero, para ver completamente si hay otros sitios de la enfermedad que no se ven en la tomografía computarizada o en la resonancia magnética (incluso para identificar tumores que pueden haberse pasado por alto en la tomografía computarizada o en la resonancia magnética) y segundo, para ver si el tumor neuroendocrino expresa un receptor llamado somatostatina. La mayoría de los tumores neuroendocrinos de grado bajo expresan el receptor de somatostatina; sin embargo, es excepcionalmente importante saber esto porque la presencia de este receptor informa al oncólogo tratante de si las opciones de tratamiento como un análogo de somatostatina (octreótido o lanreótido) o la terapia con radionúclidos del receptor peptídico son opciones. Por lo general, los oncólogos no siempre ordenan el PET con dotatate para seguimiento al largo plazo de un tumor neuroendocrino; sin embargo, es importante obtener un PET con dotatate inicialmente después del diagnóstico.
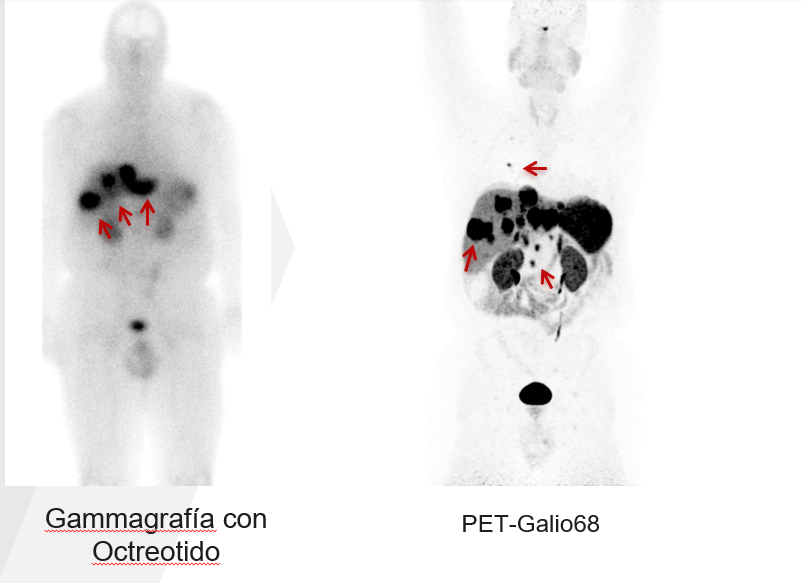
Figure 4. Comparación entre el PET-Dotatate o PET-Galio68 y el Octreoscan. PET-Galio68 puede detectar muchas más lesiones en comparación con el Octreoscan.
Expectativas (pronóstico)
Los pacientes con TNE bien diferenciados suelen tener un crecimiento lento de la enfermedad. Sin embargo, el factor que más dicta el ritmo de crecimiento del tumor es el grado de un tumor. Los TNE bien diferenciados generalmente vienen en 3 grados: grado 1, grado 2 o grado 3. Un tumor de grado 1 es el de crecimiento más lento en relación con los tumores de grado 2 y grado 3; sin embargo, los tumores de grado 1 y grado 2 generalmente se denominan tumores de bajo grado. Los pacientes con tumores de bajo grado que no se han diseminado pueden tratarse solo con extirpación quirúrgica y potencialmente pueden curarse.
El tiempo de supervivencia de los pacientes pueden variar según el grado y el estadio del tumor (local, regional (con ganglios linfáticos afectados) y distante). El tiempo medio de supervivencia de los pacientes con TNE gastrointestinales o pancreáticos de grado bajo suele oscilar entre 6 y 30 años. El tiempo medio de supervivencia de los pacientes con TNE de pulmón de bajo grado y tumores primarios desconocidos varía entre 5 y 20 años. Cuando se analizan los tiempos de supervivencia según el estadio del tumor, la supervivencia media de los pacientes con enfermedad local es de 12 a 30 años. La mediana de supervivencia de los pacientes con enfermedad regional es de 4 a 12 años. El tiempo medio de supervivencia de los pacientes con enfermedad a distancia suele ser de 2 a 4 años. Una advertencia importante es que estos tiempos de supervivencia son valores medios, no es el promedio en general, y no se aplican necesariamente a pacientes individuales. Muchos de estos valores también se basaron en datos de resultados del paciente antes de la incorporación de avances recientes en el tratamiento de TNE, como la terapia con radionúclidos receptores de péptidos (PRRT).
¿Existe una cura? ¿Qué tratamientos están disponibles?
Los TNE varían mucho en el tamaño, ubicación, síntomas y crecimiento. Por lo tanto, el tratamiento en cada caso debe individualizarse según lo que sea mejor para cada paciente de una manera individual.
La cirugía, con la extirpación completa de todo el tejido tumoral, es el primer y mejor tratamiento cuando es posible. Si los TNE se detectan temprano, no se han propagado y se eliminan por completo, esto puede resultar en una cura permanente. Sin embargo, incluso cuando no se puede extirpar todo el tejido tumoral, la cirugía puede ser necesaria para diversos fines, como el alivio de la obstrucción intestinal o el control del sangrado intestinal. A veces, en pacientes con síndrome carcinoide no controlado, la extirpación de grandes porciones del tumor (citorreducción) puede reducir eficazmente la cantidad de hormonas nocivas que se producen. Sin embargo, antes de que se considere la citorreducción quirúrgica, es importante que se discuta su caso en una junta multidisciplinaria de tumores donde hay información y recomendaciones de varios cirujanos, oncólogos médicos y radiólogos intervencionistas. Los cirujanos y radiólogos intervencionistas pueden utilizar técnicas no quirúrgicas como una sonda de congelación (crioablación) o ablación por radiofrecuencia (RFA) para destruir las metástasis de tumores neuroendocrinos en el hígado cuando no son quirúrgicamente accesibles o tumores que puedan estar cambiando de manera diferente al resto de la enfermedad. (tumores que estén creciendo cuando todos los demás sitios de la enfermedad están estables). Otra forma de eliminar el volumen de los tumores neuroendocrinos irresecables que se han diseminado al hígado es inyectar por la arteria hepática (la arteria hepática suministra sangre a las metástasis) con una combinación de material embólico (perlas) y fármacos de quimioterapia o solo con perlas. La terapia de quimioembolización interrumpe el flujo sanguíneo y el suministro de oxígeno a los tumores cuando se expone a altas concentraciones de quimioterapia que destruye el tumor y que inhibe el crecimiento. La quimioterapia en este caso se concentra en los tumores del hígado donde puede tener un efecto mucho mayor que en el resto del cuerpo. Sin embargo, la opinión experta está dividida en cuanto a si la quimioembolización es más beneficiosa que la embolización blanda sola. Otra forma de embolización es la radioembolización, que implica el uso de perlas recubiertas con sustancias radiactivas (Y90). La radioembolización se utiliza con menos frecuencia ahora que la terapia con radionúclidos receptores de péptidos (PRRT) se utiliza con regularidad para tratar a los pacientes, dado el posible riesgo de lesión hepática inducida por la radiación.
Las opciones de tratamiento sistémico para pacientes con TNE se han expandido enormemente durante la última década. Estos tratamientos varían en su capacidad para destruir células tumorales (agentes citotóxicos) versus su capacidad para congelar células tumorales (agentes citostáticos). El uso de una terapia sistémica en particular para un paciente específico es una decisión individualizada que depende en gran medida de las circunstancias del paciente.
Las opciones de tratamiento se pueden clasificar según el origen del tumor primario. Los TNE pancreáticos tienden a ser los más sensibles a la quimioterapia de todos los TNE, dado que poseen algunos defectos intrínsecos en su capacidad para reparar el ADN dañado. Uno de los regímenes que se utiliza para los TNE pancreáticos de gran volumen incluso de primera línea, es un régimen de quimioterapia oral de capecitabina más temozolomida (CAPTEM). Este régimen ha reemplazado en gran medida a los regímenes más antiguos que utilizan quimioterapia intravenosa como estreptozocina, doxorrubicina o dacarbazina. Otro régimen de quimioterapia que ha demostrado actividad en pacientes con TNE pancreáticos es el fluorouracilo más oxaliplatino (FOLFOX). Por lo general, los regímenes de quimioterapia basados ??en carboplatino o cisplatino ya no se utilizan para pacientes con NET bien diferenciados. Estos regímenes se utilizan principalmente para pacientes con carcinomas neuroendocrinos de alto grado. El régimen CAPTEM tiende a ser el más citotóxico de todos los tratamientos disponibles para pacientes con TNE pancreáticos.
Otra opción de tratamiento citotóxico para pacientes con TNE pancreáticos es la terapia con radionúclidos receptores de péptidos o PRRT. PRRT implica apuntar a los receptores de somatostatina con una partícula de radionúclido que está unida a un análogo de octreótido (descrito a continuación). El tipo de PRRT que actualmente está aprobado por la FDA para pacientes en los Estados Unidos con NET pancreáticos con expresión del receptor de somatostatina se llama Lutecio Lu 177 Dotatate (LUTATHERA®). Lu 177 Dotatate es un tratamiento intravenoso que se administra en 4 tratamientos, con cada tratamiento separado por 2 meses. Los dos principales beneficios de esta terapia son que puede reducir el tamaño de los tumores y detener su crecimiento durante un período de tiempo prolongado. Este tratamiento no está disponible en todas partes; sin embargo, tiende a estar disponible en centros grandes que se especializan en tumores neuroendocrinos. Es recomendable someterse al tratamiento en un centro que tenga mucha experiencia con este tipo de terapias. Aunque Lu 177 Dotatate es un tipo de PRRT actualmente aprobado, se están probando otros tipos de PRRT en ensayos clínicos (estos incluyen terapias alfa o la adición de sensibilizadores de radiación). Aún no se ha definido el mejor momento para usar Lu 177 Dotatate en pacientes con tumores neuroendocrinos pancreáticos.
Con respecto a los tratamientos citostáticos, varios fármacos como everolimus y sunitinib están aprobados para pacientes con tumores neuroendocrinos pancreáticos. Everolimus y sunitinib son píldoras que bloquean los factores de utilización de energía y promotores de los vasos sanguíneos para los tumores neuroendocrinos, respectivamente. Los análogos de somatostatina lanreótido y octreótido también son opciones de tratamiento para pacientes con tumores neuroendocrinos pancreáticos con expresión del receptor de somatostatina en las células tumorales. Estos agentes se administran una vez al mes como inyección subcutánea o intramuscular (en el músculo glúteo) y pueden retardar el crecimiento del tumor. Existe una formulación de acción corta de octreótido subcutáneo, sin embargo, esta se usa típicamente para pacientes con síntomas irruptivos de tumores funcionales.
El papel de la quimioterapia en pacientes con TNE no pancreáticos es menos claro. Los regímenes de quimioterapia como CAPTEM o FOLFOX pueden tener cierta actividad antitumoral en pacientes con TNE de pulmón o TNE de estómago; sin embargo, normalmente no se utilizan para pacientes con TNE del intestino delgado. A veces, para pacientes con TNE del intestino delgado de grado superior (grado 2, grado 3), se puede probar la quimioterapia. PRRT es una opción de tratamiento aprobada para cualquier paciente con NET gastrointestinal con expresión positiva del receptor de somatostatina. Muchos expertos en TNE tienden a usar esto como una terapia de segunda línea para pacientes con tumores neuroendocrinos del intestino delgado que han progresado con la terapia con análogos de somatostatina sin embargo, aún no se conoce el momento óptimo para administrar PRRT. Varios ensayos clínicos en curso (COMPETE, NETTER2) ??buscan responder a esa pregunta. Cabe señalar que, si bien la PRRT con Lu 177 Dotatate aún no está aprobada por la FDA para el tratamiento de pacientes con TNE pulmonares, a veces el seguro puede cubrir el tratamiento.
Everolimus también está aprobado para pacientes con TNE del intestino delgado y pulmón. Este fármaco retarda el crecimiento del tumor, pero normalmente no provoca una gran reducción en el tamaño del tumor. Notablemente, todavía no se han aprobado fármacos bloqueadores de vasos sanguíneos para pacientes con TNE del intestino delgado o pulmón. Esto puede cambiar en un futuro cercano con un medicamento llamado surufatinib (que está aprobado en China para tumores carcinoides y TNE no-carcinoides). La FDA aún tiene que tomar una decisión formal sobre este medicamento.
La inmunoterapia (como agente único) con inhibidores de puntos de control inmunitarios (reguladores claves del sistema inmunológico que, cuando se estimula, puede amortiguar la respuesta inmunitaria a un estímulo inmunológico) aún no ha demostrado ser muy eficaz en pacientes con TNE de bajo grado. La inmunoterapia combinada con inhibidores de los puntos de control inmunitarios puede tener más actividad en pacientes con TNE de grado superior; sin embargo, aún no se conoce la población específica de pacientes que pueden obtener el mayor beneficio de la inmunoterapia. Los tipos más antiguos de inmunoterapia, como el interferón alfa, ya no se utilizan con regularidad para el tratamiento de pacientes con TNE debido a efectos secundarios significante.
La radioterapia de haz externo en pacientes con tumores neuroendocrinos generalmente solo es útil para aliviar el dolor (particularmente cuando los tumores se han diseminado al sistema esquelético y causan dolor intenso). El tratamiento con radiación en el punto doloroso específico generalmente brindará alivio. La radioterapia estereotáctica (menos dosis con alta cantidad de radioterapia) ahora también se está explorando para tratar tumores oligoprogresivos (cuando un tumor está creciendo a pesar de que todos los otros tumores son estables) en otras regiones como el hígado.
Tratamiento de apoyo
Además de los diversos tratamientos antitumorales revisados ??anteriormente, existen muchos beneficios como resultado de una dieta nutritiva alta en proteínas, suplementos vitamínicos, en particular niacina, y suplementos minerales (como potasio, magnesio, calcio, hierro e incluso sal) cuando estos son deficientes debido a la diarrea. Es valioso reunirse tempranamente con un nutricionista después del diagnóstico para obtener orientación sobre las estrategias óptimas de dieta / suplementación. Además del uso de octreotida o lanreotida para controlar la diarrea, pueden resultar útiles los medicamentos antidiarreicos convencionales como Lomotil e Imodium. La ciproheptadina (Periactin) también puede ayudar con la diarrea y el rubor. Otro fármaco que ha sido aprobado para la diarrea por síndrome carcinoide que no responde a otros medidas o fármacos, se llama telotristat (Xermelo). Este medicamento previene la síntesis de serotonina y, por lo tanto, también puede ofrecer algunos beneficios más allá del control de la diarrea. Todos los pacientes con TNE deben tener cuidado al beber bebidas alcohólicas y evitar el estrés físico y emocional, ya que pueden precipitar ataques de crisis carcinoide. Del mismo modo, se deben evitar los medicamentos similares a la adrenalina a menos que sea absolutamente necesario. Estos incluyen varios inhaladores para el asma, descongestionantes nasales y la propia adrenalina.
Conclusión
Existe una cantidad significativa de tratamientos (actualmente disponibles y en desarrollo) para los TNE y el síndrome carcinoide, aunque la elección del tratamiento y sus aplicaciones pueden ser bastante complejas. Aunque se trata de un cáncer poco común, hay expertos disponibles que están dispuestos a ayudar y realizar investigaciones para hacer avances mediante la introducción de nuevas opciones de imágenes y terapia. Consulte la lista de expertos y otros médicos que diagnostican y tratan pacientes con tumores neuroendocrinos (tumores carcinoides y no carcinoides). See the list of experts and other physicians diagnosing and treating patients with neuroendocrine tumors (carcinoid and non-carcinoid tumors).
Como puede ver, hay buenas razones para tener esperanzas.
La Fundación del Cáncer Carcinoide, Inc. (The Carcinoid Cancer Foundation, Inc.)
118 North Bedford Road, Suite 100Mt. Kisco, NY 10549
Tel: (888) 722-3132 o (914) 683-1001
URL: https://www.carcinoid.org/
ESTE ES SITIO WEB, LAS ACTIVIDADES DE EDUCACIÓN Y CONCIENCIACIÓN Y ABOGACIA SON POSIBLES POR LAST CONTRIBUCIONES DE PARTE DE LOS PACIENTES CON TUMORES NEUROENDOCRINOS Y SUS SERES QUERIDOS DE LA FUNDACIÓN DEL CÁNCER CARCINOIDE. ¡GRACIAS!
Las preguntas generales se pueden responder llamando a la Fundación.
Cabe recalcar a nuestros visitantes que la Fundación solo puede responder preguntas generales. LA MISIÓN DE LA FUNDACIÓN ES EDUCAR SOBRE LOS CÁNCERES CARCINOIDES Y NEUROENDOCRINOS, no para realizar consultas médicas. Si se solicitara tal opinión, sería necesario revisar todos los detalles técnicos específicos en los registros médicos de un paciente, incluida la revisión de escaneos, análisis de sangre, etc. La salud de una persona es demasiado importante para tomar decisiones basadas en sugerencias casuales e impresiones generales. Se debe consultar a un médico con experiencia y experto en cáncer neuroendocrino para preguntas específicas de cada caso.
NOTA: Dado que todas las consultas reciben atención personalizada, debido a un volumen excepcionalmente grande de solicitudes de información, las respuestas por correo electrónico o por teléfono pueden demorarse un poco de vez en cuando.

